DETECTION OF IMMOBILIZED AMPLICONS BY ELISA-LIKE TECHNIQUES
Reprint from a published Paper Clinical Chemistry, 1996. Vol. 42 (9), pp. 1547-1555
Asha A. Oroskar1,*, Svend-Erik Rasmussen2, Henrik N. Rasmussen2, Søren R. Rasmussen2, Barbara M. Sullivan1,* and Arne Johansson2
1 Nalge Nunc International, 2000 North Aurora Road, Naperville, IL 60563-1796.
Fax 708/416-2519
2 Nunc A/S, Kamstrup 90, Postbox 280 - Kamstrup, DK-4000, Roskilde, Denmark
Fax + 45 46 35 0105
* Author for correspondence - Fax 708/416/2556, Telephone 708/416-2541
- Abstract
- Indexing Terms
- Non-Standard Abbreviations
- Introduction
- Material and methods
- Reagents and preparation of 5' phosphorylated and 3' radiolabelled oligonucleotides
- Temperature profiles of liquid thermal cycled in the NucleoLink wells
- Amplification by solution phase PCR in the NucleoLink strips
- Asymmetric amplification and detection of the immobilized amplicon
- Covalent binding of the solid phase primer from the Bovine Leukemia Virus (BLV) model system
- Solution phase PCR followed by asymmetric amplification
- Conversion of double stranded to single stranded covalently bound amplicons
- Detection of the single stranded covalently bound amplicon
- Optimization of the solid-phase bound PCR
- Quantitation of radiolabelled covalently bound oligonucleotides
- Results
- Covalent Binding of oligonucleotides to the NucleoLink surface.
- Thermal profiles of Liquid thermocycled in the Perkin Elmer 9600
- NucleoLink wells as traditional amplification tubes for solution phase PCR
- The Detection Of Immobilized Amplified Products in a One Phase System (DIAPOPS) or the Detection of Immobilized Solid Phase Amplicons by ELISA-likeTechniques.
- Well-to-well variation in the covalently bound oligonucleotides and DIAPOPS on the NucleoLink surface
- Asymmetric amplification on the NucleoLink surface using 5' poly-T linker modified covalently bound primers
- Signal amplification only by hybridization detection of target DNA on the NucleoLink surface
- Discussion
- Covalent binding of the 5' phosphate moiety of an oligonucleotide to the NucleoLink surface
- NucleoLink surface and asymmetric amplification
- Efficiency of solution phase PCR and detection of amplicons
- Detection of Immobilized Amplicons by ELISA-like techniques or Detection of Immobilized Amplified Products on One Phase System
- NucleoLink surface and signal amplification of the target DNA
- Other Applications of the NucleoLink surface
- Conclusion
- Acknowledgements
1. Abstract
The NucleoLink surface is a physically modified, thermostable, optically clear resin. It allows the covalent binding of the 5' terminus phosphorylated oligonucleotides. Target DNA amplification by PCR* is accomplished by asymmetric amplification on the covalently immobilized primer that develops into immobilized amplicons. A DNA fragment of the Bovine Leukemia Virus is used as a model system for the detection of immobilized amplicons by ELISA-like techniques. Covalently bound oligonucleotides are also utilized as capture probe in the hybridization-based signal amplification for detection of an infectious organism.
*PCR patents are owned by Hoffman-LaRoche, Inc., Nutley, NJ. Perkin Elmer, MJ Research, Coy and Ericomp are registered tradenames.
2. Indexing Terms
Asymmetric amplification • Immobilized amplicons • Signal amplification • Target amplification • Covalent immobilization of oligonucleotides
3. Non-Standard Abbreviations
- DIAPOPS - Detection of Immobilized Amplified Product in an One-Phase System
- BLV - Bovine Leukemia Virus
- 4-MUP - 4-methylumbelliferyl phosphate
- EDC - 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide
4. Introduction
Nucleic acid amplification has undoubtedly been the most remarkable discovery of the last decade (Saiki, R.K. et al., 1985). The simplicity of the technique is potentially conducive to the conversion of diagnostic tests from conventional immunoassay or microbiological tests to PCR-based methods. Although amplification by the polymerase chain reaction (PCR) enables at least a million fold amplification of the target DNA template, there are several issues hindering the clinical utility of PCR (Armbinder, R.R. et al., 1990; Bej, A.K. et al., 1990). Among these shortcomings, the issue of carryover contamination of amplicons has been addressed and mostly solved (Longo, M.C. et al., 1990). Presently, we are at the cross roads of implementing PCR for the routine molecular diagnosis of genetic, infectious, and malignant diseases. The use of PCR for routine molecular diagnosis is being limited by the necessity to detect the amplified product in a second phase, be it gel electrophoresis or hybridization (Keller, G.H. et al., 1988; Sauvaigo, S. et al., 1990; Landgraf, A. et al., 1992; Mahbubani, M.H. et al., 1990; Chevrier, D. et al., 1993). This two-phase assay, amplification in one reaction vessel and detection in another vessel or system (Keller, G.H. et al., 1988; Chevrier, D. et al., 1993; Cano, R.J. et al., 1993), also impedes the automation required in diagnosing multiple samples by PCR.
To offer speed, improved specificity and sensitivity, reduced contamination, and a higher throughput of sample processing, both amplification and detection need to be accomplished in one phase. Homogeneous assays, such as the TaqMan LS 50 B detection system, does offer both amplification and detection in the same liquid phase (Bassler, H.A. et al., 1995; Livak, K.J. et al., 1995). However, the amplicons in the liquid phase must still be transferred from the amplification plate to the detection plate. The ABI PRISM 7700 sequence detector on the other hand, integrates thermocycling with real-time fluorescence detection (Fox, S. et al., 1995) allowing cycle-to-cycle increases in fluorescence to correspond to standards for determining starting copy number. This procedure inherently involves the necessity of either contracting out or developing specific expertise for synthesizing fluorescently labelled oligonucleotides, a tremendous commitment to the cost of dedicated instrumentation.
A novel and cost-effective procedure that would circumvent both carryover of contaminants and increase the throughput of detection would be to have both amplification and detection coupled to a single solid phase. This entails the development of covalently bound amplicons. In order to achieve this goal, Nunc developed several proprietary surfaces. The CovaLink surface first developed at Nunc is a chemically derivatized surface (Rasmussen, S.R. et al., 1991). A “handle” with a spacer group ending in a secondary amino group is covalently grafted on to the polystyrene surface of the reaction vessel. In the presence of carbodiimide, oligonucleotides phosphorylated at the 5' terminus are bound covalently to the secondary amino group via a phosphoramidate bond.
The use of the CovaLink surface for the development of covalently bound amplicons as well as in the detection by hybridization of amplified target DNA from infectious microorganisms have already been documented (Rasmussen, S.R. et al., 1994). The CovaLink product however is not a particularly thermostable resin. Efforts were therefore focused upon the development of a second generation CovaLink-like surface with thermally stable attributes. Of these surfaces, the NucleoLink surface offers increased thermostability at temperatures typical of amplification procedures. In this study we present DNA target amplification by asymmetric amplification as well as hybridization-based signal amplification on the NucleoLink surface. In addition, several other relevant applications are also described briefly.
5. Materials and Methods
5 a) Reagents and preparation of 5' phosphorylated and 3' radiolabelled oligonucleotides
A 562-base pair fragment of the Bovine Leukemia Virus (BLV) cloned into the pGEM3Z plasmid (Promega, Madison, WI) was used as the model system for target DNA amplification by PCR (Rasmussen, S.R. et al., 1994). 5' phosphorylated oligonucleotides were obtained from several sources, including DNA Technology, Denmark; Pharmacia, Sweden; and Life Technologies, Denmark. Radiolabelled oligonucleotides were prepared by the addition of 3' [-32 P] labelled ddTTP and Terminal Transferase (Rasmussen, S.R. et al., 1991). Unincorporated radionucleotides were separated from the labelled oligonucleotide by spinning through the Spin Bind DNA Recovery System, (FMC, Rockland, ME Cat. No. 61004). Unless otherwise indicated, all chemicals used for covalent binding, washing and hybridization were obtained from Sigma Chemical Company, St. Louis, Missouri.
5 b) Temperature profiles of liquid thermal cycled in the NucleoLink wells
Six wells each from the NucleoLink strips and traditional 0.2 ml amplification tubes were filled with 50 L of water and placed directly into the Perkin Elmer 9600 thermal cycler block. Fine gauge, bare wire thermocouples were used (0.005 inch diameter wires with exposed junction) and the signal read on a digital voltmeter (A-W Sperry DM-6200). Strips were sealed during thermal cycling with a silicone rubber pad provided with the NucleoLink kit. The liquid was thermocycled through 25 cycles, each consisting of one minute dwell at 94°C, one minute at 55°C and one minute at 72°C with a pause for 5 minutes at 94°C after set point 1 in the first cycle.
The NucleoLink wells and TopYield wells (polycarbonate-based, thermoresistant resin, developed at Nunc) configured with the thermocouples were initialized during the initial 5 minutes pause at 94°C. Time `zero' was defined as the point the thermal cycler ramped from 94°C to 55°C, and the reading for each well was recorded every five seconds for approximately 4.5 minutes. The voltage was 128 VAC
5 c) Amplification by solution phase PCR in the NucleoLink strips
Solution phase PCR was conducted in a total volume of 50 L using the control DNA reagents from the Perkin Elmer (Cat. No. N801-0055)and Life Technologies PCR kits
(Cat. No. 10198-018) as recommended by the vendor.
5 d) Asymmetric amplification and detection of the immobilized amplicon
5 d i) Covalent binding of the solid phase primer from the Bovine Leukemia Virus (BLV) model system
Primers were covalently bound to the NucleoLink strips by the carbodiimide condensation reaction exactly as that described for the CovaLink NH BreakApart strips (Rasmussen, S.R. et al., 1994). To each well, 75 L aliquot was added containing one hundred ng of the 5' phosphorylated downstream primer of the BLV model system (Rasmussen, S.R. et al., 1994) dissolved in freshly made 10 mmol/L 1-methyl-imidazole (1-MeIm) pH 7.0. To achieve a final concentration of 10 mmol/L 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), 25 L of freshly made 40 mmol/L EDC dissolved in 10 mmol/L 1-MeIm pH 7.0 was added to each well. The NucleoLink strips were sealed with the Nunc Tape 8 (Nalge Nunc International, Naperville, IL, Cat. No. 249719). The NucleoLink strips were then incubated at 50°C for 5 hr. Following incubation, non-covalently bound oligonucleotides were removed using the Nunc ImmunoWasher, with 0.4 M NaOH and 0.25% Tween 20, prewarmed to 50°C. The first three washes were followed with a soak for 15 minutes at 50°C and three final washes with the same alkaline wash solution. The alkaline wash was followed with three washes of water. The precoated NucleoLink strips were either stored empty at 4°C in a sealed polyethylene bag or processed immediately for PCR. Pre-coated stored NucleoLink strips can be optionally rehydrated and blocked with 100 L of Tris-HCl pH 7.5, 150 mmol/L NaCl and 0.1% Tween 20 at room temperature for 15 minutes to avoid nonspecific binding of DNA.
5 d ii) Solution phase PCR followed by asymmetric amplification
Forty five L of PCR mix containing 10 mmol/L Tris-HCl pH 8.3, 50 mmol/L KCl, 2.5 mmol/L MgCl2,, 0.1% Tween 20, 0.015% gelatin, 200 mol/L of each dNTP, 0.5 mol/L upstream primer, 0.06 mol/L downstream primer, 5 L of varying concentrations of the Bovine Leukemia Virus template DNA (Rasmussen, S.R. et al., 1994) and 1 U Taq DNA polymerase (Life Technologies, Denmark) were placed into each well. The NucleoLink strips were sealed with Tape 8 and a silicone spacer plate provided with the kit was overlaid on the strips prior to thermocycling in the Perkin Elmer 9600 thermal cycler. The thermal cycling temperatures and number of cycles relevant for solution phase PCR were found to be appropriate for solid phase PCR. The Perkin Elmer 9600 thermal cycler was programmed for 30 cycles, each cycle consisting of three set points, 94°C for 25 sec, 55°C for 25 sec and 72°C for 10 seconds.
5 d iii) Conversion of double stranded to single stranded covalently bound amplicons
Prior to alkaline denaturation of the non-covalently bound amplicon, solution phase amplicons may be collected and electrophoresed on agarose for size identification of the amplicons. The NucleoLink strips, now emptied of the liquid phase, were washed three times with freshly made 0.2 mol/L NaOH and 0.1% Tween 20 at room temperature, soaked for 5 minutes and washed three times again in the same wash solution. Alkaline wash conditions serve to detach the non-covalently bound solid-phase amplicons. The wells were finally washed three times using the Nunc ImmunoWasher with either water or 100 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl and 0.1% Tween 20.
5 d iv) Detection of the single stranded covalently bound amplicon
Hybridization probe, biotinylated at the 5' end in 50-100 nmol/L concentration was added in 5X SSC and 0.1% Tween 20 at 100 L/well. Hybridization was carried out at 45-50°C for 1 to 20 hours. Unbound probe was removed by three washes at room temperature, soaked for 15 minutes at 50°C, and three final washes at room temperature with 0.5 X SSC and 0.1% Tween 20. A 100 L aliquot of alkaline phosphatase conjugated to streptavidin (Cat. No. D0396, DAKO, Carpinteria, CA) diluted 1:3000 in 100 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl and 0.1% Tween 20 was added to each well and incubated for one hour at room temperature. Unbound conjugate was removed by three washes, a soak for 15 minutes, followed by three washes with 100 mmol/L Tris-HCl pH 7.5, 150 mmol/L NaCl and 0.1% Tween 20 at room temperature.
An alternative protocol also gave successful detection of the hybridized biotinylated probe. Approximately 50-100 nmol/L of a biotinylated hybridization probe diluted in 6 X SSC and 5 X Denhardt's solution was hybridized at 45-50°C for 1 to 20 hours and subsequently washed with 0.1 X SSC and 0.1% Tween 20. In this scheme, the alkaline ggphosphatase conjugated streptavidin, diluted 1:3000 in Dulbecco's PBS and 0.1% Tween 20 and incubated for one hour at room temperature, was washed off first with three washes, a 15 minutes soak followed by three additional washes with PBS, 0.2 M NaCl and 0.05% Tween 20 at room temperature.
Finally, one hundred microliters of 1 mmol/L 4-methylumbelliferyl phosphate (4-MUP) dissolved in diethanolamine (pH 9.8) containing 1 mmol/L MgCl2 was added to each well and incubated for 30-60 minutes in the dark at 37-50°C.
The intensity of the fluorescent signal over the background was read in the Fluoroskan II fluorometer (LabSystems, Needham Heights, MA). Successful detection of the fluorescent signal has also been achieved with the FL 500 BioTek fluorometer (Winooski, VT).
5 d v) Optimization of the solid-phase bound PCR
The ratios of primers utilized in the solution phase were 1:8 of the covalently bound downstream primer to the non-covalently bound second or upstream primer in the BLV model system. This ratio was extrapolated from our previous studies on the DIAPOPS procedure on our CovaLink NH surface (Rasmussen, S.R. et al., 1994).
Depending upon the initial template concentration, efficient amplification on the solid-phase bound primer was found to occur with thermal cycling accomplished anywhere from 10-40 cycles in the Perkin Elmer 9600 thermal cycler, (data not shown).
5 e) Quantitation of radiolabelled covalently bound oligonucleotides
Seventy-five L of 3' 32 P-labelled 25 base oligonucleotides (0.133 ng/L; 0.266 ng/L; 0.4 ng/L; and 13.33 ng/L) in 10 mmol/L 1-methylimidazole and 25 L of freshly made 40 mmol/L EDC in 10 mmol/L 1-methylimidazole were pipetted into NucleoLink or CovaLink NH strips. Strips were incubated at 50°C for five hours. Non-covalently bound oligonucleotides were removed by washing with alkali. Wells were then dissolved in scintillation vials by overnight incubation in 100% toluene and scintillation fluid was then added to these vials. The 32 P counts were read in a 1219 Rackbeta (Wallac, Turku, Finland), liquid scintillation counter.
6. Results
6 a) Covalent Binding of oligonucleotides to the NucleoLink surface.
We analyzed the 32 P counts from covalently immobilized oligonucleotides on the NucleoLink surface. The histogram in Fig. 1 clearly demonstrates that compared with CovaLink NH, oligonucleotides bound more efficiently on the NucleoLink surface. Moreover, at lower concentrations of DNA per well, such as, 10, 20 and 30 ng/well, the binding efficiency for the NucleoLink surface was 50% but dropped to 20% when 100 ng/well were added to each well.
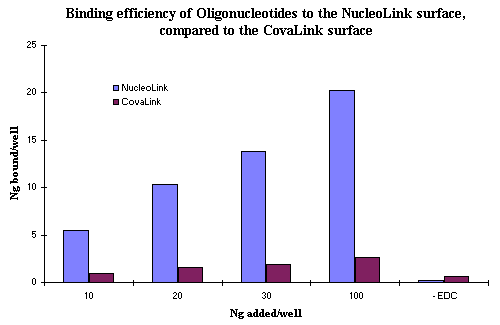
Figure 1: Binding efficiency of oligonucleotides to the NucleoLink surface. A total of 10, 20, 30, and 100 ng/well of 5' phosphorylated and 3' 32P labelled 25 base primer from the BLV system in 10 mmol/L EDC and 10 mmol/L 1-methyl imidazole was incubated for 5 hours at 50ºC in either the NucleoLink or CovaLink wells. Nonspecific or noncovalent adsorption was tested by the addition of 100 ng/well oligonucleotide in the absence of EDC. Following extensive washing with 50ºC prewarmed 0.4 M NaOH and 0.1% Tween 20, wells were analyzed for covalently bound oligonucleotides. Counts from wells dissolved in 100% toluene were read in a liquid scintillation counter. Data is presented as ng of oligonucleotide covalently bound after extensive alkaline washes. |
In a separate system, both five hour and overnight incubation with 100 ng/well, give similar binding efficiency when detected by hybridization (data not shown). Although this indicates a saturation effect, additional studies on the kinetics of oligonucleotide immobilization may provide further clarification.
6 b) Thermal profiles of Liquid thermocycled in the Perkin Elmer 9600
An average of all the six readings for the entire duration of readings taken during a specific cycle was plotted for each of the NucleoLink and TopYield strip types, as well as the Perkin Elmer thin wall tube. The comparative temperature profiles for the different groups are depicted in the graph shown in Fig. 2. The temperature profile of the Perkin Elmer tubes more closely follows that of the thermal cycler block. The superimposed curves further suggest that in both the cooling and heating phases, a temperature lag of approximately 10 seconds for the NucleoLink strips is apparent, relative to the metal block of the Perkin Elmer 9600 thermal cycler. It is important to note that despite the fact that this lag existed in the ramping phase of both the cooling from 94°C to 55°C and heating from 55°C to 72°C and also 72°C to 94°C, water temperature in both the TopYield and NucleoLink strips was relatively uniform at the set temperatures of 55°C, 72°C and 94°C for the entire one minute dwell times.
Recent experiments in the BLV system (data not shown) indicate that as short as 20 seconds may be sufficient for effective annealing for PCR. This suggests that the lag of 10 seconds in temperature equilibration may have only a marginal effect, if any, on the PCR efficiency.
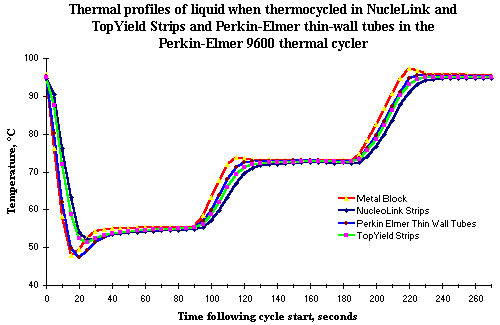
Figure 2: Thermal profiles of liquid when thermocycled in NucleoLink and TopYield strips and Perkin Elmer thin wall tubes in the Perkin Elmer 9600 thermal cycler. Fine gauge, bare wire thermocouples with `K' welded junction of 'chromel' and `alumel' were placed in wells filled with 50 L of water. Wells were sealed with a silicone rubber pad 115 x 80 mm x 3 mm thick and compared with similarly configured thin wall amplification tube and cap system from Perkin Elmer. The thermal cycler was programmed for 25 cycles, each consisting of one minute dwell at 94ºC, one minute at 55ºC and one minute at 72ºC with a pause for 5 minutes at 94ºC after set point 1 in cycle 1. The reading for each tube was initialized during the first 94ºC pause. Time zero was defined as the point the thermal cycler ramped from 94ºC to 55ºC. The reading for each well was recorded every five seconds for approximately 4.5 minutes. Every data point represents the mean of six wells which were followed individually in separate cycles. |
6 c) NucleoLink wells as traditional amplification tubes for solution phase PCR
The functional utility of the NucleoLink strips for solution phase PCR was demonstrated by the ability to amplify DNA from two different commercially available PCR kits. From the size of DNA bands evident in agarose gel photographs, Figs. 3a and 3b, it is clear that both the NucleoLink and TopYield strips may be utilized as standard solution phase PCR reaction vessels. Thus amplification of a 500 base pair fragment of DNA from the Perkin Elmer kit and a 764 base pair fragment from brain derived growth factor from GIBCO BRL PCR kit demonstrated appropriate thermal conductivity and resin compatibility with PCR for the NucleoLink surface. A comparison of the electrophoresis pattern of lambda DNA PCR products generated from the same reaction mix but thermocycled in the Perkin Elmer or the NucleoLink and TopYield strips demonstrates the functional similarity of these strips. Furthermore, no evidence of well to well contamination was evident when positive and negative samples with and without template DNA were run in adjacent wells.
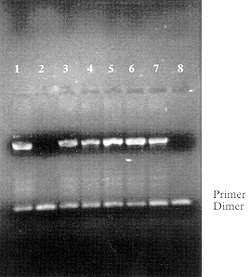 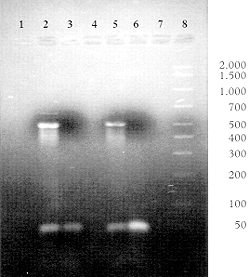
Figure 3: NucleoLink and TopYield strips as traditional amplification tubes in commercial thermal cyclers. Fifty microliters of PCR reaction mix containing reactants specified by the manufacturers were aliquoted in the specified wells of Perkin Elmer strip tubes or in the NucleoLink and TopYield strips. In each case, the filled wells were sealed first with Tape 8 and then overlaid with a silicone rubber pad 115 X 80 mm in size and 3 mm thick. The thermal cycler was programmed for a total of 40 cycles. Each cycle consisted of three set points; 94ºC for 30 sec, 55ºC for 30 sec and 72ºC for 30 sec with a 5 minutes pause at 94ºC at the end of the initial set point in the first cycle. These forty cycles were followed with a hold of 10 minutes at 72ºC. The Perkin Elmer control DNA template-primer reagents were used exactly as specified in the GeneAmp PCR reagent Kit with the AmpliTaq DNA polymerase (Cat. No. N801-0055). When Perkin Elmer strips were used, they were thermocycled separately using identical parameters as that used for the NucleoLink and TopYield strips. The agarose gel electrophoresis of amplified fragment from the DNA template is shown in Fig. 3a. With the PCR kit from Life Technologies, Inc., a DNA-primer set specific for the amplification of a 764-bp fragment, encoding a single copy of the brain-derived neurotrophic factor was tested as described in the GIBCO BRL PCR Reagent System (Cat. No. 10198-018) (see Fig. 3b). |
6 d) The Detection Of Immobilized Amplified Products in a One Phase System (DIAPOPS) or the Detection of Immobilized Solid Phase Amplicons by ELISA-like Techniques.
The DIAPOPS technique can be schematically represented as shown in Fig. 4. The entire assay performed in a single MicroWell® consists of four basic steps:
• Covalent binding of a primer to the solid phase
• Asymmetric amplification (extension of the bound primer)
• Denaturation of the bound amplicon to a single stranded fragment, and
• Detection of the bound single stranded fragment by hybridization with a labelled probe.
Addition of the PCR reagents, the DNA template and the downstream and upstream primers in ratios of 1:8 to the NucleoLink MicroWell® precoated with the downstream primer for the Bovine Leukemia Virus (BLV) model, initiated PCR in the liquid phase. Simultaneously, the DNA strand complementary to the downstream and upstream primers is amplified in the liquid phase. Hybridization of the template complimentary to the bound solid phase downstream primer, allows asymmetric extension of this primer by the Taq polymerase. At the completion of the thermal cycling, two types of amplicons, one in the liquid and the other on the solid phase are present in the primer coated NucleoLink MicroWell®. The solution-phase amplified product can either be utilized for confirmation of the presence and appropriate size of the amplified product by gel electrophoresis, or as is conventional in DIAPOPS, removed immediately after amplification, thus reducing contamination problems. The solid phase bound amplicons were converted into single stranded target DNA by washing under denaturing conditions and detected by hybridization as described in the methods section.
From the line graph in Fig. 5, it is clear that a significantly higher signal to background ratio was detected with DNA concentrations ranging from 5 X 10-14 to 5 X 10-21 moles per well of the cloned BLV template. In fact, a linear response of fluorescence was obvious for at least 4 orders of magnitude.
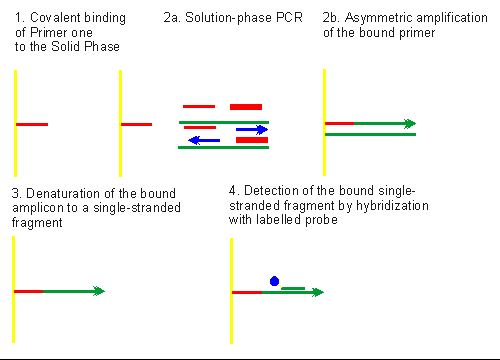
Figure 4: Schematic representation of the Detection of Immobilized Amplified Products in a One Phase System (DIAPOPS) or the Detection of Immobilized Amplicons by ELISA-like Techniques. Primer one (narrow red line; 1) is covalently immobilized in the presence of carbodiimide (EDC). Thermal cycling (see 2a) in the presence of PCR reagents, DNA template, and primers one (narrow red line) and two (broad red line) in appropriate ratios, results in the solution phase PCR amplification of the target DNA. Blue arrows indicate the direction and extension of the amplified strands in solution phase. Also, asymmetric amplification (see 2b) of the covalent solid phase bound primer leads to immobilized amplicons. Denaturation with alkali (as shown in 3) converts the solid phase double stranded amplicons to a covalently bound single stranded immobilized amplicon. Hybridization with a biotin-labelled probe (biotin label is indicated by blue circle on the probe shown in short green strip; 4) and enzymatic amplification of the hybridized complex by incubation with streptavidin-alkaline phosphatase conjugate and 4-MUP generates a fluorescent signal which is read in a microplate fluorescence reader. |
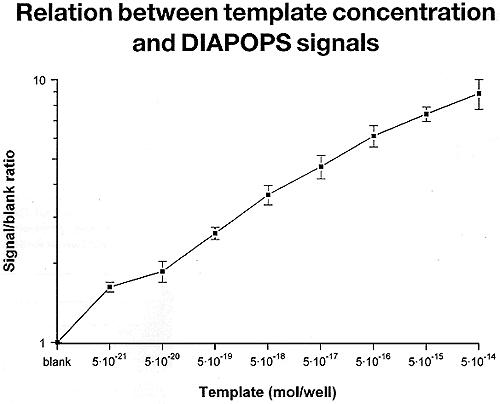 Figure 5: Detection of the immobilized amplicons (562 bp BLV template cloned in the pGEM3Z vector) with a fluorogenic substrate: relation between template concentration and the DIAPOPS signal. Asymmetric amplification and detection of the immobilized amplicons was carried out in four steps consisting of covalent binding of the solid phase primer, solid phase PCR resulting in asymmetric extension of the bound primer, conversion of the double- to single-stranded immobilized amplicon, and detection of the immobilized amplicon by hybridization with labelled probe. The biotin-labelled probe complexed with the immobilized amplicon was detected by reacting first with streptavidin-alkaline phosphatase conjugate and later with 4-MUP. The resultant fluorescent signal was read in a fluorescence plate reader, excitation wavelength 360 nm and emission wavelength at 450 nm. The results are plotted with the initial template concentration on the x-axis and the fluorescent intensities on the y-axis. Figure 5: Detection of the immobilized amplicons (562 bp BLV template cloned in the pGEM3Z vector) with a fluorogenic substrate: relation between template concentration and the DIAPOPS signal. Asymmetric amplification and detection of the immobilized amplicons was carried out in four steps consisting of covalent binding of the solid phase primer, solid phase PCR resulting in asymmetric extension of the bound primer, conversion of the double- to single-stranded immobilized amplicon, and detection of the immobilized amplicon by hybridization with labelled probe. The biotin-labelled probe complexed with the immobilized amplicon was detected by reacting first with streptavidin-alkaline phosphatase conjugate and later with 4-MUP. The resultant fluorescent signal was read in a fluorescence plate reader, excitation wavelength 360 nm and emission wavelength at 450 nm. The results are plotted with the initial template concentration on the x-axis and the fluorescent intensities on the y-axis. |
6 e) Well-to-well variation in the covalently bound oligonucleotides and DIAPOPS on the NucleoLink surface
Determination of the homogeneity of the NucleoLink surface modification and optical clarity of the resin was carried out by studies on well-to-well variation of covalently bound oligonucleotides with radioisotopic and fluorescence end-points. When eight wells coated with 5' phosphorylated and 3' biotinylated oligonucleotides were detected by binding with streptavidin-alkaline phosphatase conjugate and reaction with the 4-MUP substrate, the total C.V. calculated was found to be only 1% as seen in Fig. 6a. Six NucleoLink strips coated with 25 base oligonucleotides that were 3' end-labelled with 32 P were quantitated in a liquid scintillation counter as described in the methods section. From the standard deviations (SD) and coefficient of variation (C.V.) calculated for each of the six strips, as shown in Fig. 6b, the total C.V. was calculated to be 5.68%.
Well-to-well variation calculated for the entire DIAPOPS procedure involving oligonucleotide coating, asymmetric amplification and detection of the immobilized amplicon with a 3' biotinylated probe with streptavidin-alkaline phosphatase conjugate and 4-MUP as seen in Fig. 6c, were found to be only 12.77%.
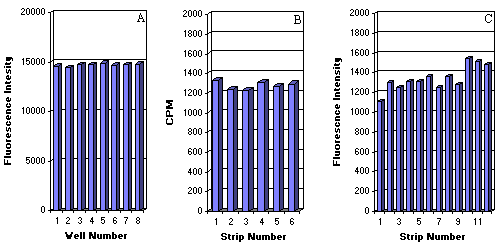
Figure 6: Evaluation of the homogeneity of the NucleoLink surface, the optical clarity of the resin for fluorescent detection, and the robustness of solid phase PCR on the NucleoLink surface. The homogeneity in the optical clarity of the NucleoLink resin was evaluated by studying the well-to-well variation in the fluorescent detection of coated oligonucleotides. A 25 base oligonucleotide phosphorylated at the 5' end and biotinylated at the 3' terminus was covalently bound to eight NucleoLink wells and detected by incubating first with streptavidin-alkaline phosphatase conjugate and later with 4-MUP. The fluorescent signal produced from 4-MUP conversion was read in the Fluoroskan II plate reader with excitation wavelength 360 nm and emission wavelength of 450 nm. From the raw counts plotted in the bar graph in Fig 6a, the mean was calculated to be 14,721.3, the SD to be 140.4 and the C.V.% to be 1%. Determination of the homogeneity of the NucleoLink surface was carried out by studying well-to-well variation in the covalent immobilization of oligonucleotides. About 100 ng/well of the 5'-end phosphorylated and 3'-end radiolabelled 25 base oligonucleotides from the Salmonella system were coated in EDC. The covalently bound oligonucleotides were evaluated as described in the methods section. 32 P -counts were analyzed from six NucleoLink strips containing eight wells each. Utilizing the mean for each strip plotted in Fig. 6b, the standard deviations and corresponding C.V.% were calculated for each strip of six. The total C.V.% was found to be 5.68%. The robustness of the DIAPOPS on the NucleoLink surface was studied by detecting well-to-well variations in the fluorescent end point obtained following coating, asymmetric amplification, hybridization with a labelled probe, and enzymatic amplification (alkaline phosphatase 4-MUP system) of the hybridization complex. Rows A through H of strips 1 through 12 were coated with primer from the Salmonella typhimurium system, processed for asymmetric amplification, hybridized with a biotin labelled probe whose signal was enzymatically amplified. Row H consisted of a negative or no DNA control. The means of fluorescence counts from rows A-G for each of the 12 strips is plotted in the histogram depicted in Fig. 6C. For each of the 12 strips the SD and corresponding C.V.% for 84 wells was calculated to be 12.77%. The mean of the blank row H is also plotted in the histogram. |
6 f) Asymmetric amplification on the NucleoLink surface using 5' poly-T linker modified covalently bound primers
In a Salmonella PCR assay, we explored the influence of the length of the covalently bound primer on the efficiency of asymmetric amplification. Amplification was analyzed from DNA purified from 5 Salmonella serotypes and from a plasmid containing the cloned target gene.
Results shown in the histogram shown in Fig. 7 demonstrated that the introduction at the 5' end of 10 thymidine residues (poly-T) on to the length of the covalently bound primer with the concurrent use of non-poly-T extended solution phase primers in ratios of 1:8 of covalently bound to non-covalently bound primer, consistently provided a higher signal to background ratio.
6 g) Signal amplification only by hybridization detection of target DNA on the NucleoLink surface
The capability of signal amplification by hybridization, without amplification of the DNA target, was determined through collaboration with the State Serum Institute in Copenhagen, Denmark. Detection of the infectious moiety with a covalently bound capture probe in a sandwich hybridization assay was carried out using a colorimetric end point. A comparison of the data obtained on the CovaLink NH and NucleoLink surfaces demonstrated the efficacy of the NucleoLink surface for target identification with covalently bound capture probes. Specifically, as demonstrated in Fig. 8, with 10 ng/well of capture probe, approximately a 10-fold higher signal to noise ratio was achieved with the NucleoLink surface as compared to that on the CovaLink surface.
Furthermore, the differences in intensities on the y-axis in the two panels indicated that a one-hour incubation with the chromogenic substrate was effective for enzymatic amplification of signal on the NucleoLink surface.
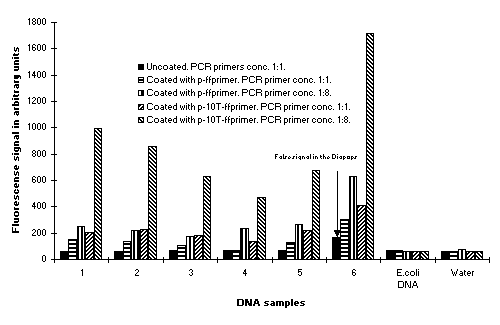
Figure 7: Modulation of the efficiency of solid phase PCR with solid phase primer extended by the addition of the 5' terminus with ten thymidine residues and variations in primer ratios. Solid phase primer extended at the 5' terminus with 0 and 10 thymidine residues (poly-T) were evaluated for the influence on the efficiencies in solid phase PCR in the Salmonella PCR assay. Data is presented on experiments where the solid phase primer 1 extended at the 5' terminus without poly-T and present in solution in ratios of unextended primer 1 to unextended primer 2 in ratios of 1:1 (pink bars) or 1:8 (red bars) was compared with the solid phase primer extended with 10-poly T and present in solution in ratios of unextended primer 1 to unextended primer 2 in ratios of 1:1 (blue bars) or 1:8 (green bars). |
7. Discussion
The NucleoLink surface is an activated surface for the covalent immobilization of oligonucleotides. A covalent bond presumably between the specified 5' terminal phosphate group on the oligonucleotide with the NucleoLink surface allows predetermined orientation and conformation of this molecule. Orientation of covalently immobilized oligonucleotides allows specified enzymatic manipulation of the molecule thus increasing its biomolecular activity. This communication presents a few of the multitude of applications that can be developed on the NucleoLink surface.
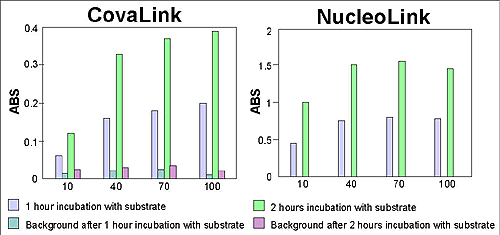
Figure 8: Signal amplification of target DNA in the NucleoLink surface. With capture probe covalently bound to the NucleoLink surface, DNA from an infectious organism was detected by a sandwich hybridization assay developed at the State Serum Institute in Copenhagen, Denmark. The hybridized complex was incubated with a labelled probe which was detected by enzymatic amplification resulting in a colorimetric end point. The color development was measured on an ELISA plate reader. Background measurements on both CovaLink and NucleoLink wells were done on uncoated strips. Note the difference in scale of the y-axes. |
7 a) Covalent binding of the 5' phosphate moiety of an oligonucleotide to the NucleoLink surface
The NucleoLink surface offers the potential to covalently bind 5' phosphorylated oligonucleotides. Through carbodiimide mediated condensation, the 5' phosphate bonds covalently to the NucleoLink surface. This chemical bond is stable against both heat as well as harsh chemicals. The resistance to chemicals allows stringent washing with 0.4 M NaOH leaving only covalently bound oligonucleotides firmly attached. This increases the subsequent specificity of the solid phase amplified products. This orientation also allows enzymatic manipulation of the 3' OH end, enabling the Taq polymerase to utilize it as a substrate in a typical PCR reaction. Since the NucleoLink resin is thermostable, the NucleoLink strips can be exposed to repeated temperature shifts or thermal cycling. With the present NucleoLink format designed to fit most 0.2 ml thermal cyclers and an optically flat, readable well-bottom, traditional PCR techniques and detection of immobilized amplicons by ELISA-like procedures is possible with the use of standard MicroWell® plate washers, plate readers and pipettes. These features make the present format of the NucleoLink strips especially suitable for asymmetric amplification and detection of the immobilized amplicons by hybridization. Given the ability to develop custom formats of the NucleoLink surface, limitless applications of covalently immobilized oligonucleotides are feasible.
7 b) NucleoLink surface and asymmetric amplification
7 b i) Efficiency of solution phase PCR and detection of amplicons
Solution phase amplified PCR products are traditionally detected by agarose gel electrophoresis. Some of the pros and cons of this detection include the ability to visualize the appropriately sized PCR product and quantitate the amount only by densitometric methods or by Southern blotting. Since a minimum of 5 ng of DNA is the detection limit on an agarose gel, approximately 7.72 X 109 copies of a 600 base pair fragment (DNA fragment from the model BLV system) that corresponds to 5 ng, are required for its detection. This high copy number needed for detection warrants that the solution phase PCR must be extremely efficient. Calculation of the efficiency states that if the detection limit should be 10 copies of target DNA and if 35 cycles are used for thermal cycling, the efficiency must at least be 0.79 in order to produce 5 ng. This is a considerably high demand on the PCR efficiency. Thus, a second amplification or more sensitive detection method is needed to detect 10 copies of target DNA. Signal amplification of immobilized amplicons by hybridization with a labelled probe circumvents the low detection limits of agarose gel detection.
7 b ii) Detection of Immobilized Amplicons by ELISA-like techniques or Detection of Immobilized Amplified Products on One Phase System
The schematic diagram presented in Fig. 4 describes the general process for asymmetric amplification on the NucleoLink surface. Generally, primers and thermal cycling conditions established for solution phase PCR can be used for asymmetric amplification on pre-coated NucleoLink wells. The solid phase primer, primer one, must still be in the liquid phase to initiate amplification. However, the optimum of primer one: primer two ratio is 1:8.
In the presence of a higher concentration of primer two, the PCR process leads to more products from primer two, resulting in an asymmetric solution phase amplification. Thus, more copies of extended primer two products are available to hybridize to the solid phase-bound primer. In reality, more covalently bound amplicons will also be produced, or asymmetrically amplified from the covalently immobilized primer one. However, analysis by agarose gel electrophoresis indicated no apparent differences in the solution phase PCR products produced in presence of primer one:two in ratios of 1:1 and 1:8 (data not shown).
Alkaline denaturation of the double stranded solid phase amplicon converts it to the single stranded form. This single stranded immobilized amplicon is amenable to detection by hybridization. The signal from this hybridization complex containing a biotinylated probe is amplified by reacting with streptavidin bound alkaline phosphatase and its substrate 4-methylumbelliferyl phosphate. The resulting fluorescent signal is a several log amplification of the signal from the original hybridization complex containing the immobilized amplicon.
The technique of asymmetric amplification and its detection on the same solid phase has several advantages over the detection of solution phase amplified products. Using the correct washing steps, the hybridization probe will only detect the covalently bound amplicons with the correct sequence, increasing the specificity of detection. Developing a clear cut-off detection limit provides a clear distinction between positive and negative samples. This can potentially lower false positives. Enzymatic amplification of the labelled probe hybridized to the immobilized amplicons enables additional signal amplification, clearly allowing a more quantitative estimate of the amplified products. Thus, 100-fold improvement of the detection limit compared to gel electrophoresis may be expected.
7 c) NucleoLink surface and signal amplification of the target DNA
Recent collaborations with the State Serum Institute in Copenhagen Denmark, on the utilization of the NucleoLink surface with precoated capture probes to detect infectious nucleic acid moieties by hybridization, underscores the general utility of the NucleoLink surface as a hybridization vessel for the detection of targets other than PCR products. DIAPOPS in the NucleoLink strips has been applied successfully for the detection of a variety of nucleic acids.
7 d) Other Applications of the NucleoLink surface
Other successful collaborations utilizing the NucleoLink surface include, the detection of Human Papilloma Virus, HPV 16 and 18 DNA cloned in a plasmid, Salmonella typhimurium DNA prepared from boiled bacterial cells, RNA from the PVY potato virus using RT-PCR in the same NucleoLink well, DNA from the bacteriophage, and DNA from the fungus, Phytophtora fragariae. Compared to gel electrophoresis, the detection limit for all the systems tested was found to be approximately 10-100 fold better.
8. Conclusion
In conclusion, the NucleoLink surface allows both nucleic acid amplification and detection on one phase. It reduces handling of the immobilized amplified products while the solution phase amplicons can be conveniently discarded. Moreover, only immobilized amplicons are detected by hybridization with labelled probe, further reducing carryover contamination by amplicons.
The NucleoLink surface allows specified surface chemistry for oligonucleotide orientation. The thermally stable resin presently in MicroWell® format with optically clear well-bottom is conducive to molding in many reaction vessel formats allowing unmatched flexibility in the laboratory automation for routine molecular diagnostics and screening.
9. Acknowledgements
The technical assistance of Sonny Madsen is gratefully acknowledged. We are indebted to the State Serum Institute, Copenhagen, Denmark for sharing their data on the signal amplification of target DNA on the NucleoLink surface. We thank Diana Krantz and Sonia Lingner, NNI Naperville, IL for their invaluable help in preparing this manuscript.